CAMI: 세체정 MAG Pipeline을 찾아서
오늘은 MAG 기반 Whole Metagenome Sequencing(WMS) 분석 pipeline에 대해 이야기해보려 합니다.
Whole Metagenome Sequencing이라는 이름만 살펴봐도 이 분석이 왜 어려운지 짐작할 수 있습니다.
일반적으로 세균의 WGS(Whole Genome Sequencing)를 진행할 때에는 하나의 균주를 분리하고, 해당 균주의 DNA를 추출해 sequencing합니다. 이후 생산된 read를 조립하여 단일 균주의 전장 유전체를 복원합니다.
그런데 metagenome은 하나의 균주만 다루지 않습니다. 하나의 sample 안에 함께 존재하는 수많은 미생물의 유전체를 동시에 관찰해야 합니다. 조금 과장해서 표현하면 한 sample 안에 있는 “겁나 많은 미생물들”의 전장 유전체를 한꺼번에 복원하려는 작업입니다.
제 짧은 경험 안에서는 sample에 존재하는 모든 미생물의 전장 유전체를 빠짐없이 복원한 사례를 아직 보지 못했습니다. 그만큼 쉽지 않은 일입니다.
얼마나 많은 데이터를 생산해야 할까
가장 먼저 부딪히는 문제는 sequencing 생산량입니다. 그리고 생산량은 곧 비용입니다.
정확한 수치는 genome 특성, read 품질, assembler, 분석 목적에 따라 달라질 수 있습니다. 다만 전장 유전체를 안정적으로 조립하기 위해 필요한 데이터량을 대략적으로 설명할 때 short-read는 약 100×, long-read는 약 30-50× 수준의 depth가 자주 언급됩니다.
예를 들어 어떤 sample 안에 200종의 미생물이 존재하고, 각 미생물의 genome size가 모두 5 Mb라고 가정해보겠습니다. 현실에서는 거의 불가능한 조건이지만, 먼저 200종이 완전히 균등한 비율로 존재한다고 가정하겠습니다.
미생물 수 = 200종
각 genome size = 5 Mb
총 genome space = 200 × 5 Mb
= 1,000 Mb
= 1 Gb
필요 sequencing yield = 1 Gb × 100×
= 100 Gb
모든 미생물의 genome을 short-read 기준 100× 수준으로 조립하려면 약 100 Gb의 clean data가 필요합니다.
하지만 실제 sample 안의 미생물은 균등하게 존재하지 않습니다. 어떤 균은 매우 많고, 어떤 균은 극히 적습니다.
같은 조건에서 가장 적게 존재하는 균의 relative abundance가 0.01%라고 가정해보겠습니다. Genome size가 5 Mb인 이 균까지 100× 수준으로 조립하려면 필요한 전체 데이터량은 아래와 같습니다.
최저 abundance 균의 목표 데이터량 = 5 Mb × 100×
= 500 Mb
필요 sequencing yield = 500 Mb ÷ 0.0001
= 5,000,000 Mb
= 5 Tb
즉, abundance가 0.01%인 미생물까지 충분한 depth로 조립하려면 약 5 Tb의 clean data가 필요합니다.
NovaSeq X와 같은 장비를 이용해 여러 run의 데이터를 생산하고 합친다면, 생산량 자체만 놓고 보았을 때 완전히 불가능한 수치는 아닐 수 있습니다. 하지만 생산된 데이터를 실제로 분석하려면 또 다른 문제가 시작됩니다.
5 Tb의 FASTQ를 분석하려면
AI에게 5 Tb 규모의 FASTQ를 분석하기 위한 대략적인 서버 사양을 물어보니 아래와 같은 답변을 받았습니다.
| 항목 | 대략적인 사양 |
|---|---|
| CPU | 128-192 threads |
| RAM | 최소 1 TB, 권장 2-4 TB |
| Scratch SSD / NVMe | 50-100 TB |
| 전체 저장 공간 | 100-200 TB |
비용도 구성에 따라 크게 달라지지만, 최소 수천만 원부터 수억 원까지 고려해야 하는 규모입니다.
물론 이 수치는 정답이 아닙니다. Sample 구성, assembler, 병렬화 전략, 중간 파일 보관 정책, cloud 사용 여부에 따라 필요한 자원은 크게 달라질 수 있습니다. 그래도 WMS 기반 MAG 분석이 단순히 FASTQ 파일을 pipeline에 넣고 기다리면 끝나는 작업이 아니라는 점은 분명합니다.
다행히 3세대 sequencing 기술인 long-read의 등장은 이 문제를 조금씩 개선하고 있습니다. Long-read는 반복 구간을 연결하고 contiguity를 높이는 데 유리합니다. Short-read만 사용했을 때보다 낮은 depth에서도 조립 가능성을 높일 수 있습니다.
물론 long-read가 낮은 abundance 문제를 완전히 해결해주는 것은 아닙니다. 그럼에도 MAG 기반 WMS 분석을 실제로 시도할 수 있는 범위를 넓혀주고 있습니다.
이러한 배경에서 MAG를 복원하기 위한 다양한 pipeline과 도구가 개발되고 발표되었습니다.
다양한 MAG 분석 pipeline과 도구
MetaWRAP, nf-core/mag, ATLAS, anvi’o, SqueezeMeta, MUFFIN, MAGNETO처럼 MAG(Metagenome-Assembled Genome)를 복원하고 분석하기 위해 개발된 pipeline은 이미 다양하게 존재합니다.
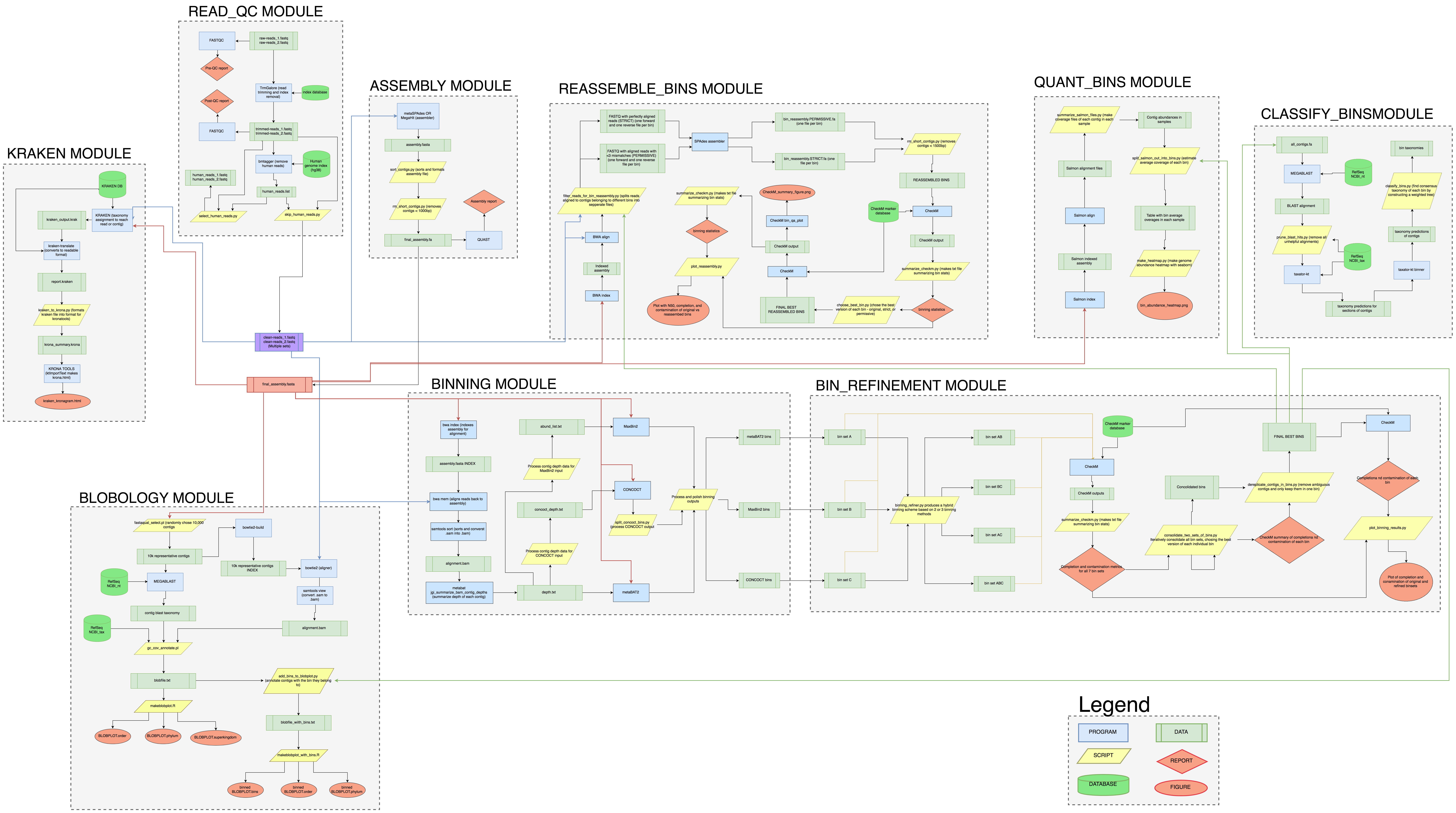
아래 그림은 MetaWRAP 논문의 supplementary material에 포함된 상세 workflow입니다. Raw read의 품질 관리부터 assembly, 여러 binner를 이용한 binning, refinement, reassembly, taxonomy 분류까지 이어지는 과정이 표시되어 있습니다.
nf-core/mag의 workflow diagram은 선택지가 더 많습니다. Short-read와 long-read 전처리 과정이 다르고, assembly, contig binning, bin refinement, quality evaluation, taxonomy 분류와 annotation 단계마다 여러 도구를 선택할 수 있습니다.
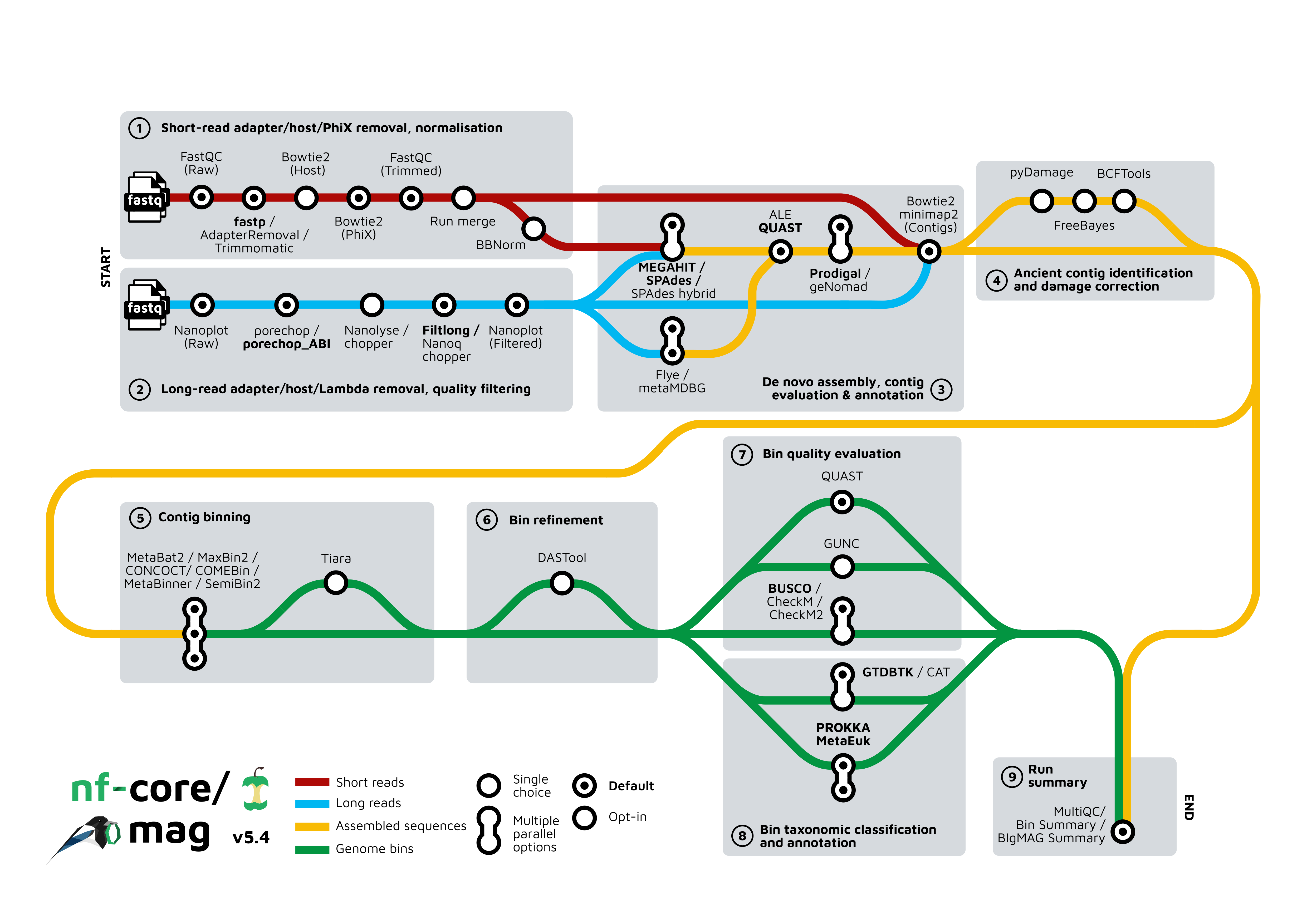
그림에서 확인할 수 있듯이, MAG pipeline은 단순히 하나의 프로그램을 실행하는 작업이 아닙니다. 입력 데이터의 종류와 목적에 맞춰 여러 도구와 옵션을 선택하고, 각 단계의 결과를 다음 단계로 연결하는 과정입니다.
전처리와 mapping 도구
fastp, FastQC, AdapterRemoval, Trimmomatic, Filtlong, Porechop, Bowtie2, BWA, minimap2, CoverM
Assembly 도구
MEGAHIT, metaSPAdes, IDBA-UD, metaFlye, hifiasm-meta, Canu, OPERA-MS
Genome binning과 refinement 도구
MetaBAT2, MaxBin2, CONCOCT, VAMB, SemiBin2, MetaDecoder, COMEBin, MetaBinner, DAS Tool, MetaWRAP Bin_refinement, Binette
MAG 평가와 taxonomy 분류 도구
CheckM, CheckM2, BUSCO, GUNC, dRep, GTDB-Tk, CAT
어떤 도구를 선택하는지에 따라 결과가 달라집니다. 같은 도구를 사용하더라도 read를 어떻게 전처리하는지, sample별 assembly와 co-assembly 중 어떤 방식을 선택하는지, binning과 refinement 단계를 어떤 순서로 연결하는지, parameter와 database를 어떻게 관리하는지에 따라 서로 다른 pipeline이 만들어질 수 있습니다.
나도 갖고 싶다, 세체정 MAG Pipeline
저도 요즘 metagenome 데이터를 이용해 MAG pipeline을 개발하고 있습니다.
분석을 업으로 삼고 비용을 받으며 일하는 Bioinformatician이기에, Pipeline을 개발하는데에 더 신중하게 되는 것 같습니다. Public으로 만들어진 유명한 Pipeline 그대로 사용하는 것만으로는 충분하지 않다고 생각합니다. 공개 pipeline은 좋은 출발점입니다. 이미 훌륭한 선배님들이 작성해 놓은 바탕으로, 검증된 도구와 분석 순서를 참고할 수 있고, 시행착오도 줄여줍니다.
하지만 실제 분석에서는 sample 종류, sequencing platform, 생산량, 분석 목적, 사용할 수 있는 계산 자원이 모두 다릅니다. 또한 실제로 이 파이프라인이 의도했을 때 보고자 하는 부분이 다를 수 있기때문에, 우리 조직에서 반복적으로 다루는 데이터에 맞춰 기존 pipeline의 장점은 가져오고, 부족한 부분은 보완해야 한다고 생각했습니다. 또한 만들어진 데이터를 어떻게 결과로써 나타내는것은 또 다른 저희만의 분야라고 할 수도 있겠습니다.
단순히 실행되는 workflow가 아니라, 결과를 설명하고 책임질 수 있는 pipeline을 만들고 싶었고, 가능하다면 기존에 알려진 pipeline보다 조금이라도 더 정확한 결과를 얻고 싶었습니다.
이번 글의 제목에 사용한 세체정은 “세계 최고로 정확한”의 줄임말입니다
아직 세계 최고라고 말할 수 있는 pipeline을 만든 것은 아닙니다. 다만 목표는 분명합니다. 우리 조직에서 만든 pipeline을 객관적인 기준으로 평가하고, 이미 알려진 pipeline과 비교하며, 하나씩 개선해보려 합니다.
그 과정에서 알게 된 프로젝트가 CAMI(Critical Assessment of Metagenome Interpretation)입니다.
내가 만든 MAG pipeline은 실제로 얼마나 정확하게 작동하는가? 를 평가해볼수 있는 좋은 Challenge를 소개하려 합니다.
![]()
1. CAMI: 정답지가 있는 모의 분석
CAMI는 metagenome 분석 도구와 pipeline을 공통 기준으로 평가하는 benchmark 프로젝트입니다.
실제 sample을 분석할 때와 Metagenome Pipeline을 검증할때 가장 답답한 점은 정답을 모른다는 것입니다. MAG가 100개 만들어져도 정말 잘 만든 것인지, 원래 있어야 할 genome을 놓친 것은 아닌지 정확히 알기 어렵습니다. 점수는 나왔는데 답안지가 없는 시험과 비슷합니다.
CAMI는 정답을 알고 있는 dataset을 제공합니다. 동일한 dataset으로 여러 pipeline을 실행하면 assembly, genome binning, virus binning, taxonomic profiling 성능을 비교할 수 있습니다. MAG pipeline을 개발하는 입장에서는 드디어 채점이 가능한 셈입니다.(기존의 Mock데이터를 이용한 Validation도 있지만 이 Mock은 실제 데이터와 달리 다양성이 너무 떨어져 검증이 충분하지 않다고 저는 생각했기 때문입니다.)
2. CAMI I: 가까운 strain을 구분하는 일은 여전히 어렵다
첫 번째 challenge 결과는 2017년 Nature Methods에 발표된 Critical Assessment of Metagenome Interpretation - a benchmark of metagenomics software에서 확인할 수 있습니다.
가장 기억에 남는 결과는 서로 유사한 strain이 함께 존재하면 복원 성능이 크게 낮아진다는 점입니다.
- 가까운 strain이 없는 genome은 비교적 잘 복원되었습니다.
- 반면 ANI(Average Nucleotide Identity) 95% 이상의 strain이 섞이면 genome recovery가 감소했습니다.
- Binning에서도 순도(purity)와 완성도(completeness)를 동시에 높게 유지하기 어려웠습니다.
- 같은 프로그램도 parameter에 따라 성능 차이가 컸습니다.
3. CAMI II: 도구는 발전했지만 문제도 어려워졌다
두 번째 challenge 결과는 2022년 Nature Methods에 발표된 Critical Assessment of Metagenome Interpretation: the second round of challenges에서 정리되었습니다.
CAMI II에서는 short-read뿐만 아니라 long-read, marine, plant-associated, strain-madness dataset까지 평가 범위가 넓어졌습니다.
Assembly 결과가 좋지 않으면 Binning도 어려워진다
Marine dataset에서 정답에 가까운 assembly 결과를 입력했을 때 binner의 평균 완성도(completeness)는 36.9%였습니다. 하지만 실제 MEGAHIT assembly 결과를 입력하면 21.2%로 낮아졌습니다. Strain diversity가 높은 strain-madness dataset에서는 5.2%까지 감소했습니다.
즉, binning 결과만 평가해서는 충분하지 않습니다. 앞 단계인 assembly 결과가 좋지 않으면 이후 단계에서 이를 완전히 보완하기 어렵습니다. MAG pipeline은 assembly부터 binning, refinement까지 전체 흐름을 함께 검증해야 합니다.
Long-read를 사용하면 항상 더 좋은 결과가 나올까?
CAMI II에서는 long-read를 함께 사용하는 hybrid assembly가 일부 지표에서 장점을 보였습니다.
- OPERA-MS는 marine dataset에서 가장 높은 연속성(contiguity)을 보였습니다.
- Hybrid assembler인 GATB와 OPERA-MS는 short-read assembler보다 16S rRNA gene을 더 완전하게 복원했습니다.
- A-STAR는 marine dataset과 strain-madness dataset에서 높은 genome fraction, 즉 원본 genome을 복원한 비율을 보였습니다.
하지만 장점만 있었던 것은 아닙니다. A-STAR는 genome fraction이 높은 대신 잘못 조립된 구간(misassembly)과 불일치(mismatch)도 더 많이 발생했습니다. OPERA-MS도 MEGAHIT보다 contiguity가 개선되었지만, long-read와 short-read를 모두 사용해 약 두 배의 데이터를 입력했습니다.
Long-read assembler인 Flye도 plant-associated dataset에서는 좋은 성능을 보였지만, marine dataset에서는 대부분의 지표에서 다른 assembler보다 낮은 성능을 보였습니다.
즉, CAMI II의 결과는 long-read가 무조건 정답이라는 의미가 아닙니다. 어떤 dataset을 분석하는지, 어떤 지표를 중요하게 보는지에 따라 적합한 assembly 전략이 달라질 수 있다는 점을 보여줍니다.
병원체를 찾는 일은 더 어렵다
Clinical pathogen detection challenge에서는 10개의 결과 중 4개가 병원체를 찾았고, 3개만이 해당 병원체를 증상의 원인으로 지목했습니다.
정확도뿐만 아니라 runtime, memory 사용량, 재현 가능성도 함께 고려해야 합니다. 결국 모든 조건에서 항상 가장 좋은 하나의 도구는 없었습니다.
4. CAMI III: 이제 직접 시험을 볼 차례
현재 CAMI에서는 세 번째 challenge인 CAMI III를 준비하고 있습니다.
본 challenge에 앞서 2026년 3월 19일에는 CAMI III Toy Human Gut dataset이 공개되었습니다. 10명의 human gut microbiome을 두 시점에서 관찰한 총 20개 sample로 구성되어 있습니다. Short-read와 long-read 데이터가 각각 총 100 Gbp씩 제공되며, gold standard assembly, genome bin, taxonomic profile도 함께 공개되어 있어 로컬에서 pipeline을 점검하기에 적당합니다.
저도 먼저 이 dataset으로 개발 중인 MAG pipeline을 검증해보려고 합니다.
- Assembly와 genome binning 결과를 정답 데이터와 비교합니다.
- Completeness, contamination, genome recovery를 기록합니다.
- Tool version, parameter, database와 사용 자원도 함께 남깁니다.
- 부족한 부분을 확인하고 pipeline 구성을 조정합니다.
5. Competition 참여를 목표로
CAMI의 FAQ를 살펴보면 challenge에 참여할 이유가 하나 더 있습니다. 신원을 공개하고 재현 가능한 결과를 제출한 참여자는 공동 CAMI 평가 논문의 저자로 참여할 수 있는 기회가 부여되는 것으로 알고 있습니다.
물론, 결과 파일만 제출하면 끝나는 것은 아닙니다. Docker container, Bioconda script 또는 설치 방법이 정리된 repository를 함께 제공해야 합니다. 출력 형식도 CAMI 표준에 맞춰야 합니다.
쉽지는 않겠지만, 그래서 더 도전해 보고 싶어졌습니다. 현재 Metagenome BI로써 나의 수준이 어느정도인지 알고싶고, 또한 이를 통해서 더 많은 공부를 할 수 있을 것이라고 생각하기 때문입니다.
먼저 Toy Human Gut dataset으로 pipeline을 검증하고, CAMI III 본 challenge가 열리면 assembly와 genome binning task에 참여해보려고 합니다. 세체정 MAG pipeline까지 갈 길은 멀겠지만, 일단 정답지가 있는 시험부터 한 번 풀어보겠습니다.
개발하고 검증하는 과정도 이후 글에서 하나씩 기록해보겠습니다.
참고 자료
- CAMI 공식 홈페이지
- CAMI FAQ
- CAMI III pre-challenge 안내
- CAMI 결과 제출 및 평가
- CAMI III Toy Human Gut dataset
- Sczyrba et al. (2017), Nature Methods
- Meyer et al. (2022), Nature Methods
-2026.06.02 by Masanam-
댓글
내용에 대한 오류 수정 요청, 질문은 언제나 환영입니다. 자유롭게 댓글을 남겨주세요! (하지만 부적절한 댓글은 삭제될 수 있습니다)